本文是上一篇帖子的进阶,仍然沿用了ChemBioOffice 2014、Gaussian 03w、Multiwfn等软件,并依然采用了Politzer拟合的参数和公式对软件计算结果进行处理,进行密度预测。不同的是本文将以Chem3D为操作平台利用Chem3D对接上的Gaussian 03w进行计算,大大节省了操作步骤,并避免了格式转换中可能出现的错误,如:单键变双键、化学键消失变孤立原子等情况。同时将B3LYP /6 –311G** 更改为B3PW91/6-31G(d,p),主要是因为Politzer处理数据拟合时采用了B3PW91/6-31G(d,p)基组,更适应拟合得到的参数。
一、Chem3D
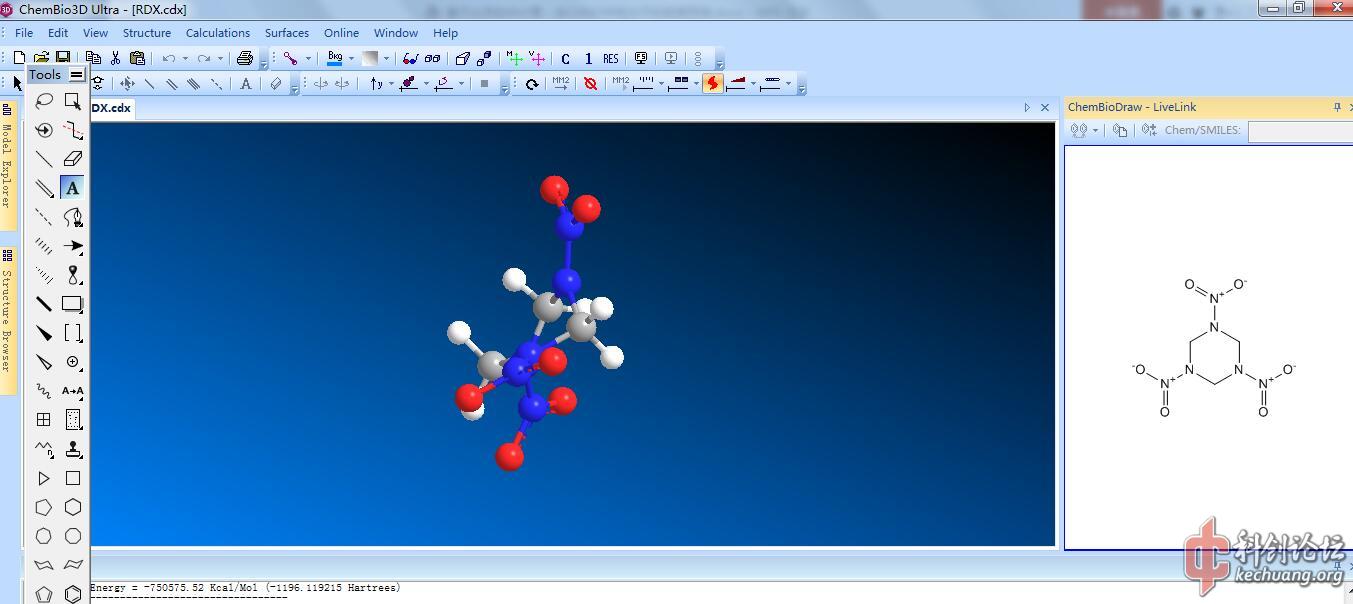
如图1.1所示,在Chem3D右侧的空白处单击鼠标左键就会弹出和ChemDraw一样的工具栏,这时按照在ChemDraw上同样的操作即可绘制出化学结构式,本文以RDX为例示范后续操作。
![1.jpg]()
图1.1 Chem3D绘图界面
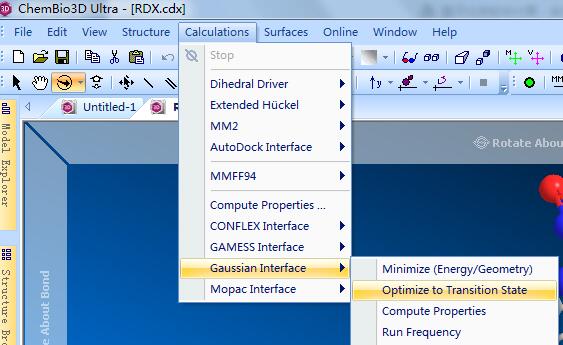
在菜单Calculations(计算)中找到Gaussian Interface(高斯接口),选择Optimize to Transition State(过渡态的优化),如图1.2所示。
![2.jpg]()
图1.2 Chem3D中高斯接口
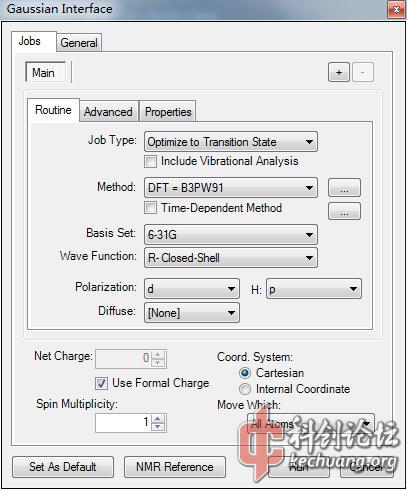
从弹出界面的Method(任务种类)中选择DFT=B3PW91,设置为6-31G(d,p)基组,如图1.3。
![3.jpg]()
图1.3 设置基组
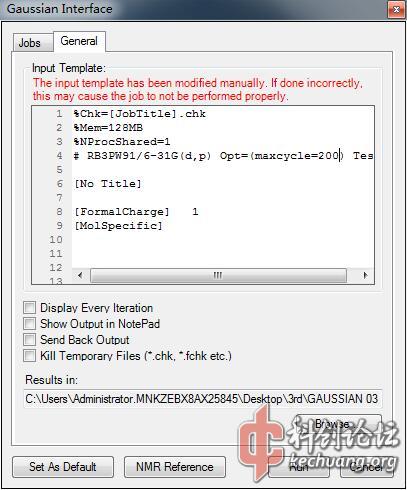
接着将opt修改为maxcycle = 200,这是为了解决L9999错误,即加大循环数来应对优化不收敛的问题。我在计算稍微复杂的分子就会出现Error termination request processed by link 9999的错误,若是平时计算完成没有错误也可以跳过这一步。
不要忘记设置fchk文件的输出路径,不然无法开始计算,而且fchk文件可直接用Multiwfn打开,不需要使用Gaussian 03将chk转换为fch文件,简化操作步骤,如图1.4。
![4.jpg]()
图1.4 输出路径的设置
采用以上方法后即可点击Run(运行)开始优化,不过以上的方法极为耗时我的联想Y480搭配i5处理器,cpu使用率50%的情况下,优化RDX耗时达3.7h。对于不想耗时那么久的朋友,可以选择Compute Properties(计算性能)任务种类,将得到的chk转换为fch文件用于计算即可,该方法一般仅耗时数十分钟,但精度较上述过渡态优化的方法稍差。
二、计算精度
由于前期计算中我没有注意到L9999错误,在优化不完善的情况下计算了十余种含能材料,浪费了大量的时间。在后期的计算中又引入了加大循环数来解决L9999错误,但随之而来的是计算时间的大大延长如出现错误时RDX加大循环前时长为0.7 h,加大循环后时长为3.7 h,在有限的时间和精力下,我只能将已完成的内容先行发表出来,后续的内容等完善后再开新帖发布。
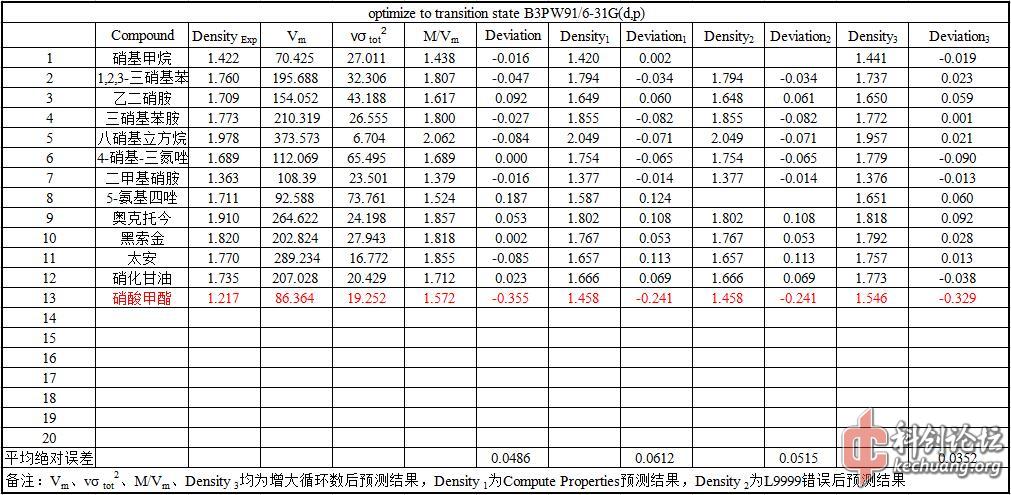
![5.jpg]()
图2.1 预测结果
文献中使用Politzer公式平均绝对误差为0.036 g/cm3,本文计算12种典型的含能材料后平均绝对误差为0.035 g/cm3,如图2.1所示。其中的硝酸甲酯结果并未计入误差计算中,因为文献中提供的硝酸甲酯密度均为标准状态下的液态密度,而本文所使用的密度预测方法预测的是晶体密度。硝化甘油的液态密度为1.596 g/cm3,固态时密度为1.735 g/cm3,本文预测密度为1.773 g/cm3,可见对于得到的结果更偏向晶体密度。
三、结论
1、本文仅计算得到了12个有效数据,而文献中则计算了36个含能材料。所以本文计算得到的平均绝对误差与文献中略有出入。
2、使用过渡态的优化的任务类型进行密度预测得到的结果精度较高,但极为耗时。在处理复杂结构或需大批量处理数据时可选择计算性能任务类型,该方法平均绝对误差为0.049 g/cm3,依然有着一定的精度。
3、出现L9999错误后的数据等效于计算性能任务类型得到的数据,二者预测的密度一致。
4、在计算了脂肪族硝基类、芳香族硝基类、脂肪族硝胺类、氮杂环硝胺类、氮杂环硝基类、无氧氮杂环类、笼型硝基类、硝酸酯类7个类型的含能材料后,对比实测密度,平均绝对误差仅0.035 g/cm3,符合文献中计算了36个含能材料后平均绝对误差0.036 g/cm3,精度较高可用于新型含能材料设计工作中。
5、数据上仍与文献有出入,在本文方法还有着提升空间。





200字以内,仅用于支线交流,主线讨论请采用回复功能。